Predicting pathogenic variants (in genetic diseases & breast cancer)
Predicting the pathogenicity of variants of uncertain significance (VUS) in BRCA is critical for estimating the risk of hereditary breast and ovarian cancer (HBOC).
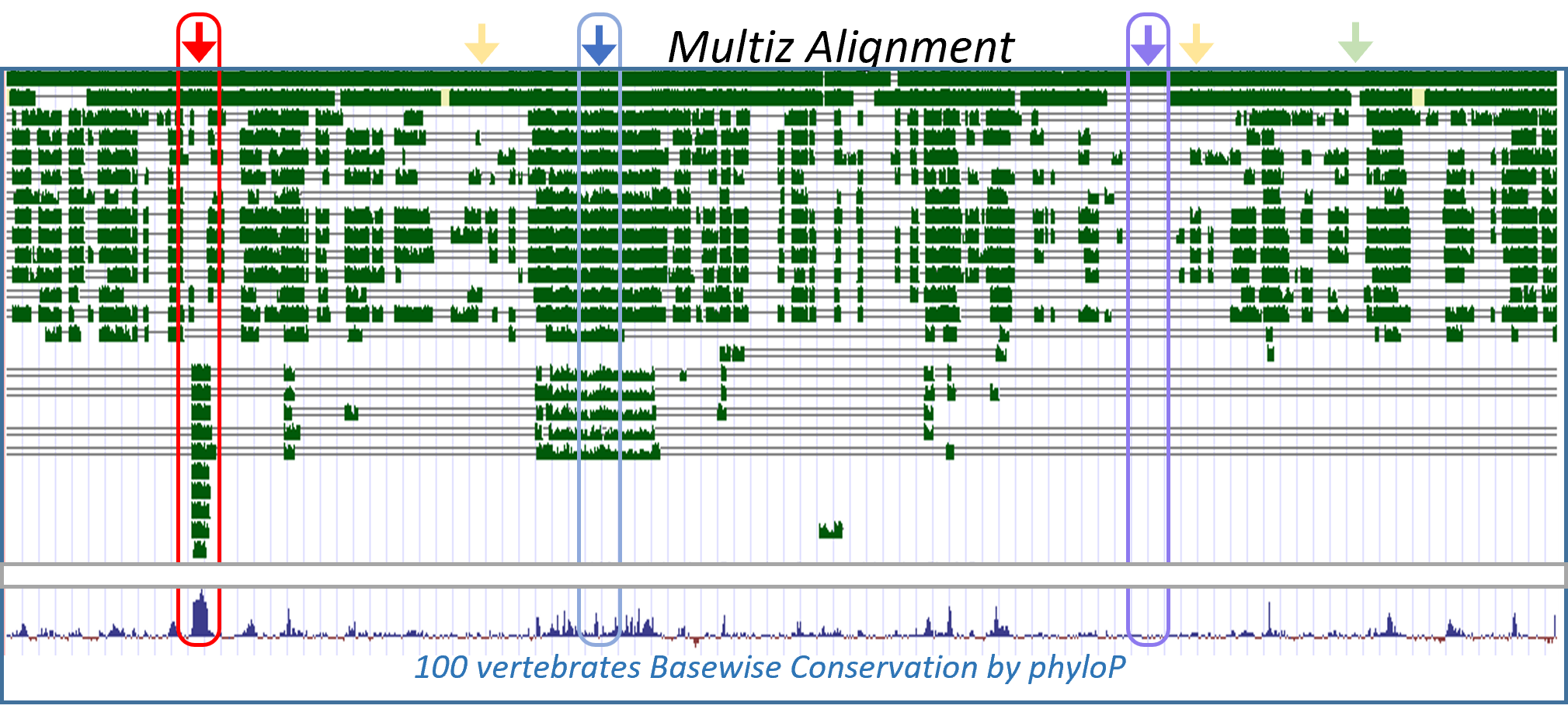
High level of conservation at a genomic position is a strong predictor for the pathogenicity of variants in this position. However, single nucleotide variants (SNVs) are frequently located at positions with complex conservation patterns, where the nucleotides are sporadically conserved across vertebrates. The meaning of these patterns, their variability among genes, and their association with variant pathogenicity were never assessed.
Here we analysed the conservation patterns of SNVs in 115 disease-associated genes that include BRCA genes, across 99 species, to extract additional information from conservation data.
We developed EvoDiagnostics, a random forest-based model that uses nucleotide conservation patterns and outperforms baselines in predicting variants in BRCA1 (AUC-0.925), BRCA2 (AUC-0.930), and in the entire variant pool of the 115 disease-genes (AUC-0.933). We found that the pathogenicity of variants is better learned from their complex conservation patterns, compared to naïve conservation, and that the conservation of some species is more informative than others in the context of specific genes. Our work characterizes conservation patterns and their variability among genes and species, and highlights the significance of conservation patterns in variant prioritization.
EvoDiagnostics could be either used as a stand-alone prediction tool or as a complementary measurement for ensemble prediction methods.